Болестта на Кройцфелд-Якоб е рядко невродегенеративно заболяване, което бързо, постепенно и силно засяга мозъка.
Кройцфелд-Якобска болест (CJD) постепенно унищожава мозъчните клетки и причинява малки дупки в мозъка. Хората с CJD ще имат атаксия или трудност при контролиране на движенията на тялото, необичайна походка, реч и деменция.
Тя винаги е фатална и няма лечение.
CJD засяга една личност на всеки милион глобално всяка година, включително Съединените щати (САЩ).
Причините могат да бъдат спорадични, наследени или придобити. Това засяга най-вече хора на възраст над 60 години и това е рядко при хора под 30-годишна възраст.
Лице с CJD е малко вероятно да оцелее повече от една година след появата на симптомите.
Какво представлява CJD?
![[CJD]](https://demedbook.com/images/1/what-is-creutzfeldt-jakob-disease-cjd.jpg)
CJD е трансмисивна спонгиформна енцефалопатия (ТСЕ), която разрушава мозъка във времето. То се причинява от инфекциозен агент, наречен прион. Приопът не е вирус или бактерия.
Други видове ТСЕ включват синдром на Gerstmann-Sträussler-Scheinker (GSS), фатална фамилна безсъние и куру на хора. Други примери са скрейпи при овце и кози, спонгиформна енцефалопатия по говедата (СЕГ) или "болест на луда крава" при говеда.
Подобни енцефалопатии и синдроми на отслабване се откриват при други видове и те са показали, че са преносими при лабораторни животни.
Центровете за контрол и превенция на заболяванията (CDC) отбелязват, че класическата CJD не е свързана с СЕГ или други варианти.
Симптоми
CJD има дълъг инкубационен период. Симптомите могат да отнеме до 40 години. Симптомите се появяват, когато мозъчните клетки се унищожават. Състоянието на пациента ще се влоши бързо в рамките на седмици, като повечето хора умират в рамките на една година.
Характерните симптоми на CJD са бърза прогресия към деменция и миоклонус, или спазматично принудително движение на мускулни групи.
Промените в настроението или поведението, промените в личността, загубата на памет и влошената преценка са често срещани. Условието може да прилича на деменция от Алцхаймер или на болестта на Хънтингтън, но симптомите ще се развият в рамките на дни до седмици, а не години.
Тъй като болестта прогресира, проблемите с координацията и миоклонът се влошават, а зрението се влошава, което води до слепота. В крайна сметка пациентът вече не може да се движи или да говори и те ще навлязат в кома.
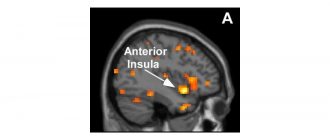
Аутопсиите на мозъчната тъкан разкриват, че CJD включва някои уникални промени, които не се наблюдават при други деменции.
Има няколко варианта на CJD, които не са задължително свързани с класическата CJD, а симптомите и хода на заболяването може да са различни.
Причини
CJD се случва, когато прион протеин, анормален вид амилоиден протеин, причинява аномалии в други протеини. Натрупването и малформацията на приони върху мозъчните клетки в крайна сметка водят до увреждане на мозъка и смърт.
То може да бъде спорадично, наследено или придобито.
142214
Спорадични CJD
В 85% от случаите CJD е спорадичен. Няма очевидни рискови фактори.
Наследствено CJD
Между 5% и 10% от случаите са наследени. Те се появяват, когато възникне промяна в гена, който контролира образуването на прион протеини. Може да има фамилна анамнеза за CJD, или може да възникне мутация в яйцеклетките или в клетките на сперматозоидите, поставяйки потомството в риск от развитие на болестта.
Прионите не съдържат генетична информация и не се нуждаят от гени, които да се възпроизвеждат, но мутацията в гена за нормалния прионов протеин на организма може да доведе до ненормални действия на прионите.
Няколко различни мутации в прион гена са идентифицирани. Специфичната мутация, установена във всяко семейство, засяга колко често се появява заболяването и кои симптоми са най-забележими.
Не всеки с мутации в гена на прион протеин развива CJD.
Придобита CJD
Няма данни, че всеки тип CJD може да се предава от едно лице на друго, но някои процедури са свързани с предаването на CJD.
Те включват:
- трансплантация на роговицата
- електроди импланти
- бъбречна трансплантация или менингеална присадка, която е присадка на външното покритие на мозъка
- използването на човешки растежен хормон
Около 1% от случаите се предават чрез известно или много подозирано излагане на засегната тъкан на мозъка или на нервната система.
Спонгиформна енцефалопатия по говедата
През 90-те години един вид CJD е свързан с излагане на СЕГ, което се случва при говеда.
![[прионна теория]](https://demedbook.com/images/1/what-is-creutzfeldt-jakob-disease-cjd_2.jpg)
Счита се, че предаването е свързано с консумацията на храна. Този вариант е имал тенденция да се отразява на по-младите пациенти и продължил по-дълго.
СЕГ засяга редица видове, включително едър рогат добитък, хора и котки.
Някои учени смятат, че необичаен "бавен вирус" или друг организъм причинява CJD, но все още не са изолирали конкретен вирус или организъм при хората с тази болест.
Агентът, който причинява CJD, има няколко характеристики, които не са обичайни за вирусите и бактериите.
Това включва дълъг инкубационен период, факта, че е трудно да се убие и че не изглежда да съдържа никаква генетична информация под формата на нуклеинови киселини, ДНК или РНК.
Учените смятат, че CJD и други ТСЕ не са причинени от жив организъм, а от приони. Прионите не са живи, но те са протеини с необичайни структури, които се разширяват в мозъка.
Това разширяване уврежда мозъчната тъкан и причинява характерните симптоми на CJD.
диагноза
Няма тест за потвърждаване на диагнозата на CJD. Само биопсия на мозъка може да направи това и това е твърде рисковано за пациента, докато е жив.
Тестовете могат да ви помогнат да намерите най-вероятната причина.
Физическото изследване ще търси мускулни спазми и ще проверява рефлексите на пациента. Те могат да бъдат по-реактивни от нормалното. Мускулите могат да бъдат прекалено тонизирани или изсъхнали, в зависимост от това къде болестта засяга мозъка.
Визуалният или очния тест може да открие частична слепота, която пациентът не е забелязал преди това.
Електроенцефалограмата (ЕЕГ) може да разкрие необичайни електрически импулси.
КТ или MRI може да изключи инсулт като причина за симптомите.
А лумбална пункция, или гръбначен кран, може да тества гръбначния флуид, за да изключи други причини за деменция. Той може да покаже дали има инфекция или повишено налягане в централната нервна система (ЦНС).
Ако протеинът 14-3-3 се намира в течността и човекът показва типични симптоми, има голям шанс за това, че лицето има CJD.
Мозъчните биопсии след смъртта показват, че мозъчната тъкан е гъста, с видими малки дупки, където са съсипани купчини от нервни клетки.
лечение
Няма лечение за CJD, и никакви лекарства не могат да помогнат за контрола или забавянето на прогресията на заболяването.
Лечението има за цел да облекчи симптомите и да направи пациента възможно най-удобен.
Опиатните лекарства могат да помогнат за облекчаване на болката. Клоназепам и натриевият валпроат могат да помогнат за облекчаване на неволни движения, като например мускулни потрепвания.
В по-късните етапи пациентът трябва често да се движи, за да предотвратява леглото. Катетърът може да се използва за източване на урината и храненето е чрез интравенозни течности.
Предотвратяване
Превантивните мерки включват стерилизация на цялото медицинско оборудване, за да бъдат унищожени всички организми, които могат да причинят заболяването, и да не се допуска дарение от роговица от хора с анамнеза за диагноза или възможно CJD.
Повечето страни вече имат строги указания за управление на заразените крави и ограничения относно фуражите, за да се избегне възможността за предаване на други форми на ТСЕ на хората.
Хората, които са изложени на лица с диагноза CJD, трябва да следват някои насоки.
Те включват:
- покриващи отворени рани, разфасовки и ожулвания върху кожата
- носещи ръкавици при работа с тъкани, кръв или течност на пациента
- носещи рокля за еднократна употреба или дрехи за контакт с пациент
- като използвате защитен капак за лице, защита на очите или маска, когато съществува опасност от пръскане на замърсена течност
- стерилизиращо оборудване, което е било използвано върху или близо до пациента
Извършва се проучване на ролята на прионите в CJD, за да се установи как болестта засяга мозъка и да се намери ефективно лечение.