Мускулната атрофия на гръбначния стълб е генетично заболяване, при което човек не може да контролира движението на мускулите си, поради загуба на нервни клетки в гръбначния мозък и мозъчния ствол.
Загубата на мускулите и слабостта водят до затруднение да стоят, да ходят, да контролират движенията на главата и дори в някои случаи да дишат и да преглъщат.
Спиналната мускулна атрофия (SMA) не е само едно условие, а редица заболявания. Групирани заедно, различните видове SMA формират втората водеща причина за нервно-мускулна болест.
SMA присъства при 1 на всеки 6 000 до 10 000 живородени. Това е второто най-често срещано фатално автозомно рецесивно разстройство след кистозна фиброза.
Ако SMA засяга бебе на възраст под 2 години, то обикновено е фатално. Ако пациентът е по-възрастен при появата на заболяването, той ще има различен тип SMA и очакваната продължителност на живота може да е нормална.
Няма лекарство за SMA, но през декември 2016 г., първото лекарство за лечение на това е одобрено: Spinraza.
Бързи факти за спиналната мускулна атрофия:
Ето някои ключови въпроси за спиналната мускулна атрофия (SMA). По-подробно е в основната статия.
- Има различни видове SMA. Те се различават по тежест.
- Това е генетично състояние.
- Основният симптом е мускулна слабост и загуба на мускули. В тежки случаи може да възникнат респираторни проблеми.
- Леченията включват поддържащи устройства, които помагат на пациента да диша, и ново лекарство, Spinraza.
- SMA не може да бъде предотвратено, но бъдещите родители могат да поискат генетични тестове, ако те могат да бъдат носители.
Видове
Има различни видове SMA. Те се различават по отношение на кога започват да се появяват в индивида и в очакваната продължителност на живота, която човекът с разстройството ще има.
SMA тип I

SMA тип I е сериозно състояние. Децата с това разстройство никога не успяват да седят или да застанат. Обикновено е фатален преди навършване на 2 години.
Тя може да бъде открита преди раждането, тъй като може да има намаляване на движението на плода през последните месеци на бременността. Ако не, това ще стане очевидно в рамките на първите няколко месеца от живота.
Бебета с типа SMA Никога не седя или не, а те обикновено не преживяват до двегодишна възраст. SMA тип I е известен също като болест на Werdnig-Hoffmann.
SMA тип II
SMA тип II обикновено се появява на възраст между 3 и 15 месеца. Децата могат да се научат да седят, но никога няма да могат да стоят или да ходят.
Продължителността на живота зависи от това дали пациентът развива респираторни проблеми. Повечето хора със SMA тип II оцеляват до зряла възраст.
SMA тип III
SMA тип III или болест на Kugelberg-Welander, се появява на възраст между 2 и 17 години.
Симптомите включват необичайна походка и трудност при движение, катерене или изкачване от стол. Възможно е да има лек треперене на пръстите. Някои хора могат да загубят способността си да ходят и те също могат да развият сколиоза. Усложненията включват затлъстяване и остеопороза.
Синдром на Кенеди
Синдромът на Кенеди е известен също като прогресивна мускулна атрофия със спинобулбар. Синдромът на Кенеди е бавно прогресивно наследено състояние, което обикновено се проявява между 20 и 40 години, но може да се появи по-късно.
Жените носят гена, но само син ще наследи разстройството.
Вродена SMA с артрофиппозис
Вродената SMA с артрокрипсия е рядко заболяване. Хората с това състояние ще имат трайна контрактура на ставите, известна като артрокрипсия.
Състоянието е очевидно при раждането. Характеристиките включват тежки контрактури, кривина на гръбначния стълб, деформация на гръдния кош, респираторни проблеми, необичайно малка челюст и увиснали горни клепачи.
Възрастен SMA
Възрастните SMA или SMA IV започват след 18-годишна възраст. Хората с това състояние могат да ходят и нямат проблеми с дишането или храненето.
Изследователите са открили връзки между SMA и амиотрофичната латерална склероза (ALS), често наричана болест на Lou Gehrig.
Симптоми
Симптомите на SMA зависят от неговата тежест и от възрастта на лицето, когато започне. Деца със SMA тип I се раждат с много малък мускулен тонус, слаби мускули и проблеми с храненето и дишането. При SMA тип III, симптомите може да не се появят до втората година от живота.
Във всичките му форми основната характеристика на SMA е мускулната слабост, придружена от атрофия на мускулите. Това е резултат от деневриране или загуба на сигнала за договаряне, който се предава от гръбначния мозък.
Този сигнал обикновено се предава от моторните неврони в гръбначния мозък до мускулите чрез аксона на моторния неврон. В SMA, или моторният неврон с аксон, или самият аксон, става нефункциониращ. Тя спира да работи.
Много от симптомите на SMA се отнасят до вторични усложнения на мускулната слабост. Те могат да бъдат облекчени отчасти чрез терапия.
Причини
SMA се случва, когато моторните неврони в гръбначния мозък и мозъчната става не работят или спират да работят поради генетични промени. Моторните неврони са нервните клетки, които контролират движението.
Всяка човешка клетка съдържа част, която получава инструкции от гените и когато инструкциите съдържат грешка, това се нарича изтриване. Частта, която получава инструкциите обикновено е протеин.
В SMA, инструкциите, които се дават на моторните неврони или нервите, които контролират движението, съдържат изтриване, което причинява недостиг на протеини. Генът, отговорен за инструкциите за моторните неврони, е моторизиращ неврон, обикновено SMN 1.
Всеки човек има 2 двойки гени за всяка дадена инструкция. Един ген се наследи от майката и един от бащата. Някои заболявания ще се появят, ако само един от наследените гени съдържа грешка в инструкцията. Други заболявания, като SMA, ще се появят само ако има грешка както в наследствените гени на майката, така и на бащата.
За да има дете SMA, и двамата родители трябва да дадат SMN 1 с погрешни инструкции.
Въпреки това, дори и двамата родители да имат дефектния ген, детето не винаги ще го наследява. Дори сред тази популация шансът е само 1 на 4 на бременност, че детето ще има SMA. Един от 40 възрастни са носители на гена, който причинява SMA.
диагноза
Диагнозата обикновено започва, когато родителите или болногледачите забелязват симптоми на SMA при дете.
Лекарят ще извърши подробна медицинска история, семейна история и физически преглед. Те ще видят дали мускулите са флопи или нестабилни, за да проверят за дълбоки сухожилни рефлекси и мускулна фасцикулация на мускулите на езика.
Тестовете, използвани за диагностициране на SMA, включват кръвни изследвания, мускулна биопсия, генетични тестове и потенциално електромиография (ЕМГ). ЕМГ се използва за оценка на здравето на мускулите и нервните клетки или моторните неврони, които ги контролират. Изследването на амниоцентезата или хорионния вил може да направи оценка на плода по време на бременността.
лечение
Все още няма лечение за SMA и няма начин да се предотврати това, тъй като това е наследствено условие. Лечението обаче може да помогне на хората да живеят по-пълноценен живот.
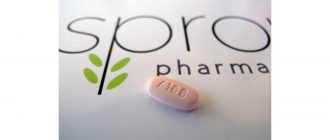
Spinraza
През декември 2016 г. Американската администрация по храните и лекарствата (FDA) одобри лекарство nusinersen (Spinraza) за лечение на SMA. Това е първото лекарство, което трябва да бъде одобрено за това състояние.
Тя се дава чрез инжектиране, първите три дози на 14-дневни интервали, четвъртата след 30 дни, а след това на всеки 4 месеца.
Spinraza насочва основния дефект в SMA, така че може да помогне за забавяне, предотвратяване или дори обръщане на симптомите. Честите нежелани реакции включват по-висок риск от инфекция на дихателните пътища и запек. Възможно е също да има риск от кървене и бъбречни проблеми.
Помощни устройства
Помощните технологии като вентилатори, електрически инвалидни колички и модифициран достъп до компютри позволяват на хората с SMA да живеят по-дълго, да бъдат по-активни и да участват в общността.
Вентилацията е особено важна. Тежестта на слабостта на индивида пряко засяга хода на заболяването. Кърмачетата с тежка SMA могат да получат респираторни заболявания, тъй като мускулите, които поддържат дишането, са слаби.
Децата с по-леки форми на SMA могат да очакват по-дълъг живот, въпреки че може да се нуждаят от богата медицинска помощ.
Молекулярната биология е подобрила нашето разбиране за SMA. Провеждат се много експериментални лечения, включващи генна замяна, замяна на стволови клетки на моторни неврони и терапии за увеличаване на експресията на ген SMN2.
SMA е генетичен и няма начин да го предотвратим.
Родителите с семейна история на SMA се насърчават да търсят генетично консултиране, преди да започнат семейство.